
Le coronavirus du syndrome de détresse respiratoire aiguë sévère (SRAS-CoV-2) pénètre dans les cellules humaines à l’aide de sa protéine de pointe qui se lie au récepteur de l’enzyme de conversion de l’angiotensine 2 (ACE2) de l’hôte. La réaction protéine de pointe-membrane hôte dépend du clivage protéolytique, ainsi que de l’activation des glycoprotéines de l’enveloppe virale, par les protéases de la cellule hôte. Les protéases notables à cet égard comprennent la protéase transmembranaire, la sérine 2 (TMPRSS2), la cathepsine L (CTSL) et la cathepsine B (CTSB).
Les thérapies disponibles aujourd’hui ciblent soit différentes étapes du cycle de vie viral (analogues nucléotidiques ou antiviraux à large spectre), soit le système immunitaire de l’hôte (médicaments immunosuppresseurs ou anticorps monoclonaux), soit les lésions vasculaires aiguës (médicaments antihypertenseurs et anticoagulants). En raison de leur rôle dans l’initiation de l’infection virale, le récepteur ACE2 et TMPRSS2 peuvent être les cibles thérapeutiques les plus cruciales.
À ce jour, les antirétroviraux ont été au centre de la plupart des recherches thérapeutiques, suivis par les médicaments anticancéreux (par exemple, les inhibiteurs de kinases) et les antimicrobiens en tant que prochains candidats médicaments les plus prometteurs. En raison du coût élevé et du temps requis pour le développement de médicaments de novo, le repositionnement des médicaments apparaît comme une option viable.
Le criblage traditionnel à haut débit (HTS) consiste à tester des milliers à des millions de petites molécules en parallèle. Les « hits » HTS permettent d’identifier des cibles thérapeutiques et de valider des effets biologiques même lorsque l’on sait peu de choses sur le composé. Cependant, cela a un coût substantiel en termes de temps et de ressources, nécessitant le criblage de bibliothèques de centaines de milliers de petites molécules pour obtenir quelques composés actifs (0,01 % – 0,14 %) pour une enquête plus approfondie. Le dépistage virtuel peut surmonter ces problèmes dans les premières phases du développement d’un médicament. De plus, la ressemblance avec un médicament ou uneabsorption, rédistribution, mmétabolisme, excrétion, et tdes critères d’oxicité (ADMET) peuvent être intégrés au processus pour augmenter encore la qualité des candidats sélectionnés.
Des stratégies bioinformatiques basées sur des données provenant d’analyses omiques multiples, des mécanismes d’action et différentes altérations moléculaires de la maladie à coronavirus (COVID-19) ont été proposées pour le repositionnement de médicaments et l’identification de nouveaux médicaments pour COVID-19 ont été proposées. D’autre part, des stratégies de chimioinformatique, basées sur la modélisation quantitative de la relation structure-activité (QSAR) et l’amarrage moléculaire ont également été utilisées par les chercheurs pour cribler des cibles thérapeutiques.
Des chercheurs ont récemment publié une étude dans la revue Briefings en bioinformatique dans lequel ils ont intégré plusieurs méthodes bioinformatiques et chimiques pour prioriser les médicaments pour le traitement de COVID-19.
Détails de l’étude
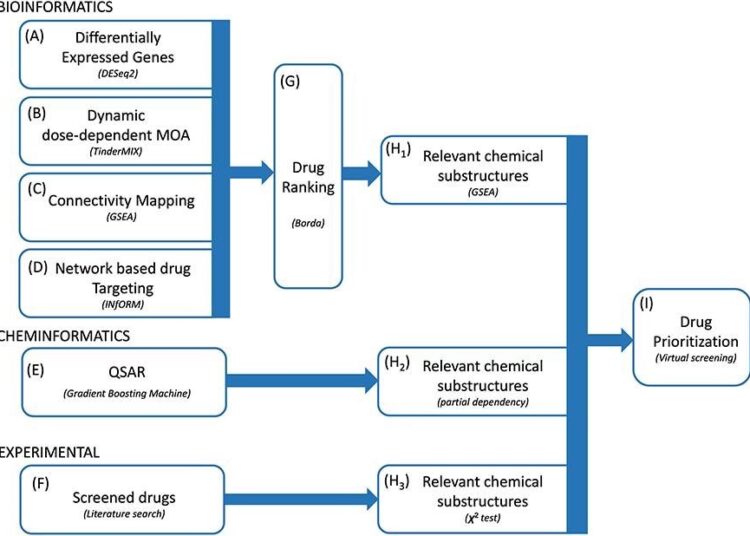
L’ensemble du cadre d’étude a été conçu pour fonctionner avec quatre approches bioinformatiques complémentaires. L’hypothèse principale derrière ces approches était que les gènes topologiquement centraux dans le réseau ont un rôle central dans l’adaptation à l’exposition au virus. Par conséquent, les chercheurs ont priorisé les médicaments en fonction de l’importance de leurs cibles génétiques dans le réseau. Les chercheurs ont ensuite identifié un classement robuste des médicaments utilisant la méthode Borda et extrait une liste de sous-structures chimiques pertinentes.
Méthodologie proposée. Nous avons intégré plusieurs méthodes bioinformatiques et chimioinformatiques pour prioriser les médicaments pour le traitement de COVID-19 (A-E). Notre cadre se compose de quatre approches bioinformatiques complémentaires, y compris l’analyse d’expression différentielle (A), le MOA dynamique dose-dépendant (B), la cartographie de connectivité (C) et le ciblage de médicament basé sur un réseau (D) ainsi qu’une méthode de chimioinformatique basée sur QSAR ( E). Nous avons en outre complété notre ensemble de sous-structures chimiques candidates avec celles extraites de médicaments actifs testés expérimentalement dans plusieurs études (F). Les quatre approches bioinformatiques sont fusionnées pour trouver un classement robuste des médicaments (G). A partir du rang produit par les approches bioinformatiques, la méthode QSAR et de la liste des médicaments criblés, trois listes de sous-structures chimiques sont identifiées (H1-H3) dans le but d’augmenter la robustesse des prédictions ainsi que de générer des connaissances facilement utilisables. dans le cadre du développement de nouveaux médicaments. Finalement, nous avons exploité l’ensemble des sous-structures chimiques candidates en effectuant une analyse de criblage virtuel de la base de données DrugBank (I).
D’autre part, les chercheurs ont utilisé des méthodes chimiques basées sur QSAR pour identifier les sous-structures chimiques de médicaments prédictifs du niveau de dérégulation du récepteur ACE2, de la protéase transmembranaire TMPRSS2 et des enzymes protéolytiques de surface cellulaire CTSB et procathepsine L (CTSL).
Sur la base de la présence de ces sous-structures chimiques, une liste de 700 médicaments candidats efficaces contre le COVID-19 a été identifiée à partir de la base de données DrugBank parmi 8 000 autres. Les critères d’inclusion étaient un compromis entre la sélection d’un ensemble de médicaments qui représentaient le mieux le haut de la liste des priorités, en fonction de leurs sous-structures chimiques, et plusieurs considérations pratiques telles que le prix, la disponibilité, le temps d’expédition et la facilité de stockage. Les chercheurs ont validé leur méthode en réalisant une évaluation biologique in vitro de 23 médicaments sélectionnés en tenant compte de ces critères.
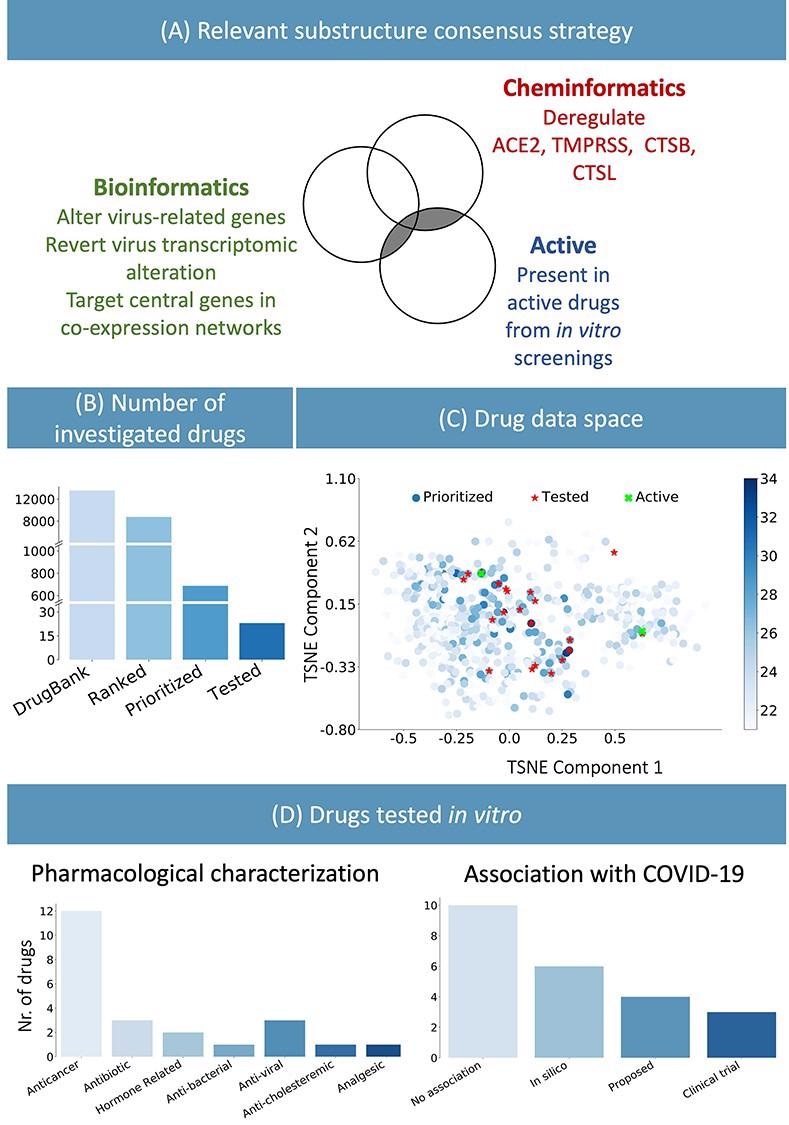
(A) Stratégie de consensus pour identifier la sous-structure chimique pertinente, en utilisant des méthodes bioinformatiques et chimico-informatiques ainsi que des résultats expérimentaux de la littérature publiée. (B) L’approche suggérée permet de réduire le nombre de tests expérimentaux : l’ensemble de la base de données DrugBank a été filtré à moins de 800 médicaments pertinents et des tests in vitro ont été effectués sur 23 candidats. (C) Représentation graphique des médicaments prioritaires. La nuance bleue représente le nombre de sous-structures chimiques identifiées en (A), présentes dans les médicaments. Les 23 composés sélectionnés sont affichés en rouge. Ils ont été sélectionnés parmi les médicaments partageant la sous-structure la plus pertinente et répondant à des critères logistiques pratiques. Sur les 23 médicaments, les deux surlignés en vert ont été identifiés expérimentalement comme actifs. (D) Caractérisation pharmacologique et description de l’association connue avec COVID-19 des 23 médicaments testés. In silico fait référence aux médicaments dérivés d’études in silico, tandis que proposé fait référence aux médicaments suggérés pour leur rôle thérapeutique potentiel dans la littérature.
12 des 23 médicaments identifiés étaient des thérapies ciblées en oncologie, et 8 d’entre eux étaient des inhibiteurs de kinase. Semblables aux médicaments antiviraux, les médicaments anticancéreux peuvent également cibler des processus biologiques qui jouent un rôle crucial dans la modulation de la réponse immunitaire, la division et la mort cellulaires, la signalisation cellulaire et la génération de microenvironnement dans le système hôte.
Les chercheurs ont découvert que deux médicaments, la 7-hydroxystaurosporine et le bafetinib, ont montré une inhibition significative de l’infection virale. De plus, l’analyse des images des cellules infectées par rapport aux cellules traitées a montré que la formation de cellules syncytiales multinucléées était également significativement réduite. De manière inattendue, lorsqu’ils sont combinés, les deux médicaments ont exercé une inhibition synergique encore plus puissante de l’infection virale ainsi qu’une inhibition de la fusion cellule-cellule à des concentrations plus faibles. Davantage in vitro des expériences ont montré que les médicaments combinés étaient efficaces même après une heure d’infection des cellules. Cela suggère que la combinaison pourrait avoir entravé un mécanisme de post-entrée du virus. De plus, les résultats ont également confirmé l’efficacité de la combinaison des médicaments contre la variante delta plus infectieuse du SRAS-CoV-2.
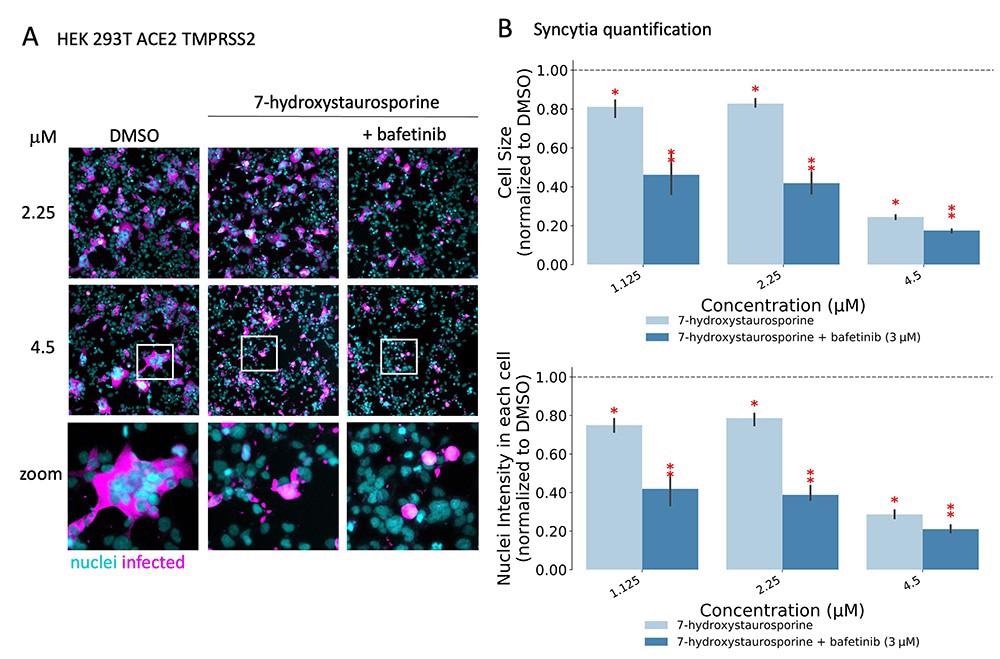
La 7-hydroxystaurosporine et le bafetinib inhibent la syncytie induite par le virus. (A) Images de fluorescence représentatives de cellules HEK-293 T-AT traitées avec les médicaments indiqués 1 h avant l’infection. Cellules fixes 16 hpi ; cyan = noyaux, magenta = cellules infectées. Les zones agrandies de chaque image sont indiquées par des cases blanches. (B) Quantification de la taille des cellules et du contenu nucléaire de l’expérience en (A); valeurs normalisées à la médiane des contrôles DMSO. Toutes les valeurs représentent les moyennes de trois expériences. Les barres d’erreur indiquent la SD. Les astérisques rouges indiquent des valeurs p significatives (<0,05) pour le test t unilatéral entre chaque traitement et le DMSO.
Implications
La présente étude a démontré un débit élevé et une méthode efficace pour modéliser des médicaments spécifiques au COVID-19, ainsi que pour sonder des cibles thérapeutiques. De plus, cette étude a montré que la 7-hydroxystaurosporine et le bafetinib combinés avaient le potentiel de contrôler efficacement les dommages du SRAS-CoV-2. En plus de fournir une première ligne de référence dans la gestion des situations d’urgence, il serait également important pour les futures recherches sur les thérapeutiques contre le COVID-19.
















