Un nouvel ensemble de données mondial de 239 virus à ARN infectieux pour l’homme montre comment les hôtes animaux, la transmission vectorielle, les lacunes en matière de surveillance et les caractéristiques virales façonnent le chemin allant de la propagation à la menace épidémique.
Étude : Un catalogue complet de virus à ARN infectieux pour l’homme. Crédit d'image : Andrzej Rostek/Shutterstock
Une revue récente publiée dans la revue Données scientifiques présente un catalogue mondial mis à jour qui apporte le nombre d'acide ribonucléique (ARN) virus connus pour infecter les humains chez 239 espèces, soit 25 de plus qu'en 2018, offrant de nouvelles informations sur l'émergence et la propagation.
Plutôt que d’apparaître au hasard, la plupart des virus se regroupent au sein de quelques familles, sont liés à des hôtes non humains, en particulier des mammifères, et sont détectés à des rythmes variables au fil du temps, à mesure que la taxonomie, la déclaration, la surveillance et les technologies de séquençage évoluent.
Bien que les retombées sur l’homme soient courantes dans le monde entier, seule une minorité d’espèces atteint des niveaux épidémiques ou endémiques chez l’homme, mettant en évidence un goulot d’étranglement critique entre l’exposition et la propagation de l’épidémie.
ARN Les virus restent aujourd’hui une menace croissante pour la santé mondiale, entraînant des maladies telles que la rougeole, la grippe et le sida causées par le VIH et provoquant de nouvelles épidémies. Les événements récents impliquant le virus Oropouche et le coronavirus 2 du syndrome respiratoire aigu sévère (SRAS-CoV-2) soulignent le potentiel épidémique de ces virus. Pourtant, le paysage viral continue d’évoluer rapidement.
Les chercheurs identifient de nouvelles espèces infectieuses pour l’homme presque chaque année, révisent les classifications et élargissent les données génomiques et écologiques. À mesure que les preuves s’accumulent sur la transmission, la gamme d’hôtes et la propagation, la nécessité d’un catalogue mis à jour devient essentielle pour suivre ce qui est connu et anticiper les risques futurs.
Sommaire
Méthodes du catalogue des virus à ARN humain
Dans cet article, les chercheurs ont développé un ensemble de données actualisé et étendu sur ARN virus connus pour infecter les humains, capturant les connaissances actuelles jusqu’en décembre 2024.
S'appuyant sur des catalogues antérieurs de 2001 et 2018, ils ont mené des recherches documentaires systématiques tous les 1 à 3 ans en utilisant des bases de données telles que Web of Science, PubMed, Scopus et Google Scholar, complétées par des sources telles que l'Organisation mondiale de la santé (OMS), Centres de contrôle et de prévention des maladies (CDC), ProMed et les enregistrements génomiques du National Center for Biotechnology Information (NCBI).
L'ensemble de données comprenait uniquement des rapports primaires évalués par des pairs fournissant des preuves solides que ARN virus reconnus par le Comité international pour la taxonomie des virus (TICV) infecter les humains dans des conditions naturelles ou réelles, à l'exclusion d'une inoculation expérimentale intentionnelle ou in vitro preuve.
L’équipe a résolu les ambiguïtés grâce à des évaluations indépendantes et à un consensus et, dans certains cas, a déduit des traits manquants de virus étroitement apparentés. Ils ont compilé des données au niveau des espèces en intégrant des informations sur les sous-types connus et ont lié chaque virus à son premier cas humain signalé, à sa séquence génomique et à son origine géographique.
Les chercheurs ont enregistré les caractéristiques clés, notamment la transmissibilité, la gamme d’hôtes et les voies de transmission, à l’aide de critères standardisés. Ils ont classé la transmissibilité en niveaux 2, 3 et 4, allant des infections zoonotiques sans propagation humaine aux virus capables de se propager de manière épidémique ou endémique chez l'homme.
Enfin, l’équipe a cartographié les dates et les lieux de découverte, permettant ainsi des analyses temporelles et spatiales de l’émergence du virus. En intégrant les données génomiques, écologiques et épidémiologiques dans un cadre unique, l'ensemble de données mis à jour fournit une base solide pour étudier la diversité virale, l'évolution et les risques pour la santé publique.
Le décompte annuel des espèces de virus à ARN infectieux pour l'homme, nouvelles et (actuellement) reconnues par l'ICTV, est présenté à la figure 2a. La figure 2b représente l'accumulation d'espèces au fil du temps, ainsi que l'accumulation de genres et de familles contenant une ou plusieurs espèces de virus à ARN infectieux pour l'homme. Le premier virus humain à ARN – le virus de la fièvre jaune – a été signalé en 1901. Le nombre d’espèces augmente lentement jusqu’au milieu des années 1950, puis un peu plus rapidement. À la fin du 20e siècle, 178 espèces avaient été identifiées, et jusqu'à présent, au 21e siècle, 61 autres ont été ajoutées. Par décennie, les années 1960 ont marqué le plus grand nombre de nouvelles espèces (42). Ce fut ensuite dans les années 2000 (31), mais le taux baissa à nouveau dans les années 2010.
Modèles mondiaux de découverte de virus à ARN
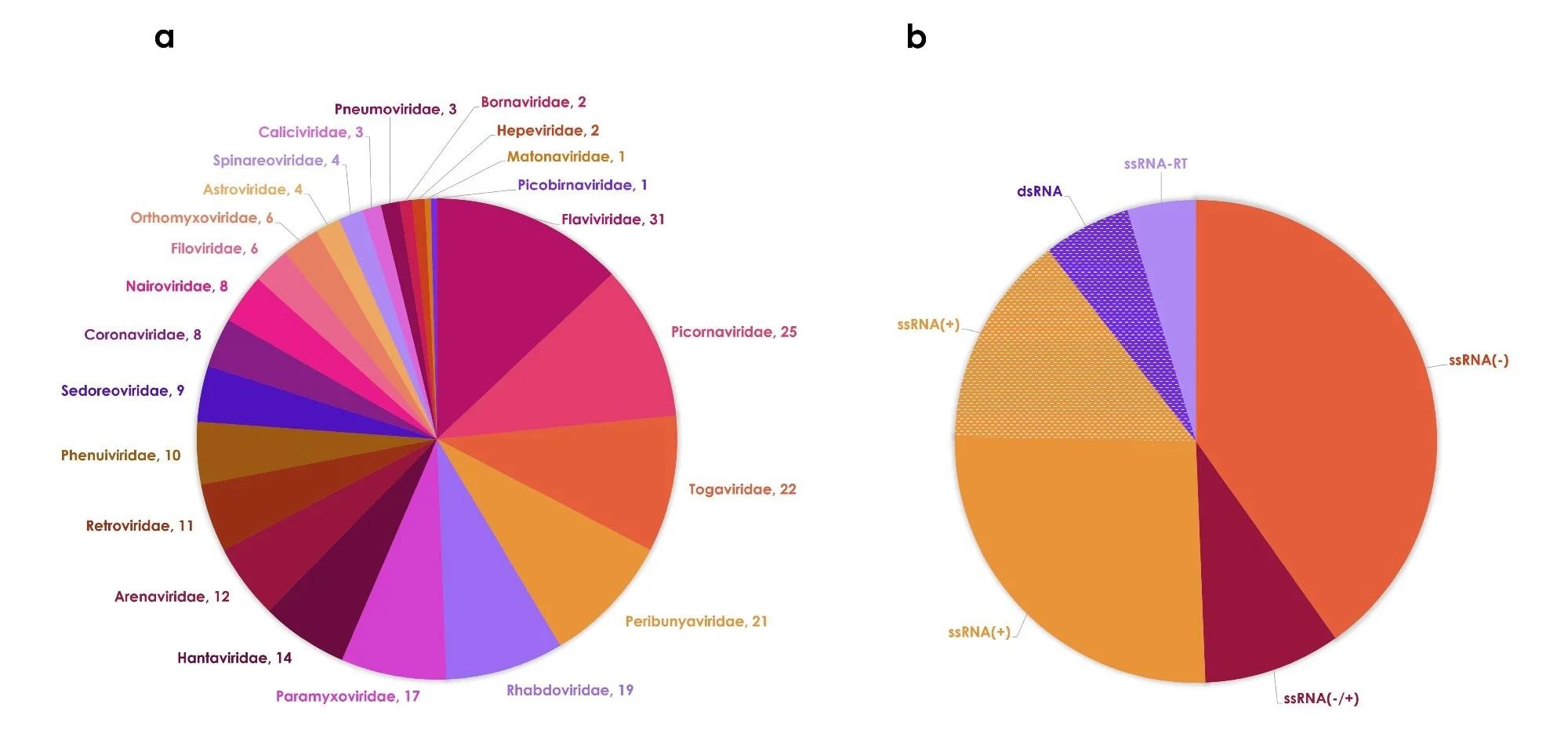
L'ensemble de données mis à jour comprend 239 ARN virus connus pour infecter les humains, tels que classés par le TICV. Par rapport à 2018, cela reflète 25 espèces supplémentaires identifiées grâce à de nouvelles découvertes et mises à jour taxonomiques.
Ces espèces couvrent 61 genres et 23 familles, bien que la diversité reste concentrée dans quelques familles et que la plupart des virus partagent des caractéristiques génomiques communes, en particulier monocaténaires. ARN génomes.
Au fil du temps, les découvertes ont augmenté à partir du milieu du 20e siècle, même si les auteurs notent qu'une analyse formelle est nécessaire pour déterminer si les taux de découvertes augmentent ou diminuent globalement.
Après une croissance minime au début du XXe siècle, les taux d’identification ont fortement augmenté à partir du milieu des années 1900, avec des sommets notables dans les années 1960 et au début des années 2000. Cependant, la plupart des espèces nouvellement identifiées étendent les genres et familles existants plutôt que d’introduire des groupes taxonomiques entièrement nouveaux.
Géographiquement, les premiers cas humains signalés se sont produits sur tous les continents habités, avec des clusters dans les régions dotées de systèmes de surveillance plus solides. Cette tendance met en évidence à la fois la nature mondiale de la propagation virale et l’influence de la capacité de détection sur la découverte.
Débordement, transmission et potentiel épidémique
Sur le plan écologique, la majorité des virus (62 %) sont strictement zoonotiques (niveau 2) et ne supportent pas la transmission interhumaine. Seules 60 espèces atteignent le niveau 4, ce qui signifie qu’elles sont endémiques chez l’homme ou susceptibles de se propager épidémiquement, et nombre d’entre elles conservent encore des réservoirs animaux.
La plupart des virus sont associés à des hôtes mammifères non humains, renforçant ainsi leur rôle central dans l’émergence. Les voies de transmission sont diverses, mais la propagation vectorielle, principalement via les moustiques et les tiques, domine, suivie par les voies d'inhalation et de contact direct.
Les voies de transmission d’un sous-ensemble de virus restent notamment incertaines, reflétant des lacunes persistantes dans les connaissances. Ensemble, ces résultats mettent clairement en évidence un paysage défini par des retombées documentées répétées, des découvertes croissantes et une adaptation limitée à une transmission humaine durable.
Surveillance des virus à ARN et prévision des risques
Ces résultats suggèrent une approche plus ciblée et proactive face aux menaces virales émergentes. Plutôt que de rechercher à grande échelle des agents pathogènes entièrement nouveaux, les efforts futurs pourraient utiliser cet ensemble de données pour examiner les familles virales à haut risque, les réservoirs de mammifères et les régions à surveillance limitée, où les retombées non détectées sont les plus susceptibles de se produire.
L’expansion du séquençage génomique, de la métagénomique et de la surveillance en temps réel sera essentielle pour combler les lacunes persistantes dans les connaissances, en particulier en ce qui concerne les voies de transmission et la gamme d’hôtes.
Dans le même temps, l’ensemble de données constitue une base précieuse pour modéliser les tendances en matière de découverte et identifier les traits liés au potentiel épidémique. À mesure qu’elle continue d’évoluer, elle peut aider à affiner la prévision des risques et à orienter les systèmes d’alerte précoce.
En fin de compte, le défi n’est pas seulement de découvrir de nouveaux virus, mais aussi de comprendre lesquels sont les plus susceptibles de s’adapter, de se propager et de constituer la prochaine menace sanitaire mondiale.