
Une nouvelle étude publiée sur le serveur de préimpression bioRxiv * en juin 2020 montre que la proportion d'appariement A / T dans les génomes viraux peut augmenter la tendance à infecter les humains en raison des caractéristiques moléculaires correspondantes de certains gènes hôtes, ce qui augmente la sensibilité de l'hôte au virus.
L'épidémie de COVID-19 qui a commencé à Wuhan, en Chine, s'est maintenant propagée dans le monde entier, infectant plus de 10 millions de personnes et causant plus de 500 000 décès. La biologie et la propagation du virus font l'objet d'un examen scientifique approfondi depuis lors, étant donné l'urgence de stopper la propagation du virus avec un vaccin ou un antiviral efficace.
Sommaire
Correspondance homme-virus
Le syndrome respiratoire aigu sévère coronavirus 2 (SARS-CoV-2) appartient à la famille des coronavirus (CoV) que l'on trouve chez de nombreuses espèces hôtes. Le virus antérieur du syndrome respiratoire aigu sévère (SRAS) a été étudié pour comprendre la voie de propagation et les réservoirs d'animaux potentiels pour de tels agents pathogènes.
Début 2007, les scientifiques étaient parvenus à la conclusion que les chauves-souris hébergeaient de nombreux nouveaux virus capables de franchir les barrières d'espèces pour infecter également les êtres humains, mais également de nombreux virus étroitement liés au SARS-CoV. Ces virus étaient connus pour être très variables, ce qui explique le plus grand risque qu'ils présentent pour les humains et les autres animaux domestiques par rapport à d'autres virus
Le risque de transmission zoonotique augmente le risque d'émergence de ces maladies en cas de commerce fréquent d'espèces sauvages dans le monde. L'hypothèse sous-jacente est que si un virus est capable de se répliquer dans plusieurs hôtes, il s'adapte dans un compromis entre une correspondance précise et fonctionnelle afin qu'il puisse s'adapter à la large gamme de molécules d'ARNt dans différents hôtes. D'un autre côté, un seul hôte nécessitera des gènes viraux spécialisés.
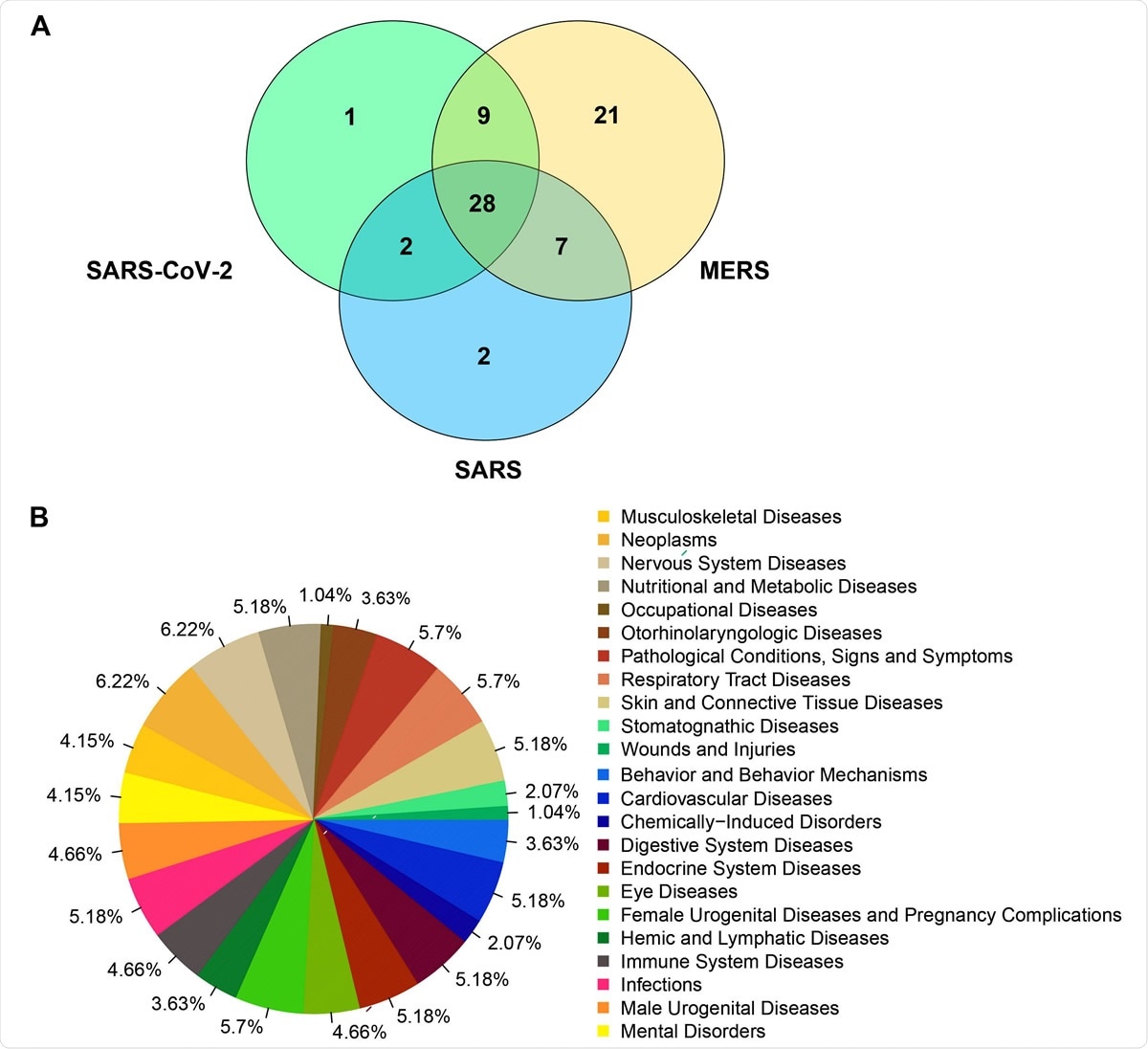
, le SRAS (NC_004718) et le MERS (NC_038294) en fonction des caractéristiques moléculaires.B). Fréquences des maladies associées aux gènes humains regroupés avec les gènes viraux du SRAS-CoV-2, du SRAS et du MERS dans l'analyse de regroupement.")
Diagramme de Venn représentant le nombre de gènes humains qui se sont regroupés avec les gènes viraux pour le SRAS-CoV-2 (NC_045512), le SRAS (NC_004718) et le MERS (NC_038294) en fonction des caractéristiques moléculaires.B). Fréquences des maladies associées aux gènes humains regroupés avec les gènes viraux du SRAS-CoV-2, du SRAS et du MERS dans l'analyse de regroupement.
L'étude: Identifier le couplage moléculaire SARS-CoV-2-humain
L'étude actuelle menée par deux chercheurs de l'Université de Buenos Aires vise à identifier les modèles moléculaires viraux et les codons préférés, qui reflètent la machinerie des cellules hôtes et sont, par conséquent, la structure génique virale préférée pour une viabilité virale optimale et une sensibilité humaine. Les scientifiques ont suggéré que le biais des paires de codons et les préférences des dinucléotides sont les principaux facteurs qui reflètent l'utilisation des codons de l'hôte, comme cela a été prouvé avec les études d'atténuation du virus par désoptimisation des paires de codons.
Les scientifiques avaient le double objectif de découvrir la nature moléculaire et adaptative des trois principaux CoV humains ainsi que de découvrir les facteurs des cellules hôtes qui ont sélectionné les codons viraux. Deuxièmement, ils ont tenté d'identifier les gènes viraux essentiels à la réplication et les gènes humains nécessaires au même processus. Cela aidera à décider si la variabilité de la population dans le contenu génétique modélise les caractéristiques des gènes et aide à développer la sensibilité de l'hôte.
Les chercheurs ont examiné environ 500 séquences de génomes téléchargées à partir du NCBI, y compris le SRAS, le MERS et le SARS-CoV-2, classés par l'hôte. En utilisant uniquement des génomes de référence, la qualité a été évaluée et les virus représentatifs ont été sélectionnés pour une analyse plus approfondie.
Les chercheurs ont ensuite sélectionné 463 gènes hautement exprimés dans le tissu pulmonaire des hôtes humains, avec au moins une différence entre leur niveau d'expression ici et dans le tissu avec le niveau hautement exprimé suivant.
Analyse du biais d'utilisation des codons
Ils ont effectué une analyse du biais d'utilisation des codons (CUB) en utilisant le contenu GC total du CDS ainsi que celui des première, deuxième et troisième positions de codon, notées respectivement P1, P2 et P3. Ils ont également calculé les indices de codon tels que l'utilisation relative des codons synonymes (RSCU), le nombre effectif de codons (ENc), l'indice d'adaptation des codons (CAI), l'indice de biais des codons (CBI), la fréquence optimale des codons (Fop), la moyenne générale Hydropathicité (GRAVY), aromaticité (Aromo) et contenu en GC aux première, deuxième et troisième positions de codon (GC1, GC2 et GC3), fréquence d'un G ou C à la troisième position de codon de codons synonymes (GC3) , la moyenne de GC1 et GC2 (GC12) et la sélection translationnelle (TrS2).
À l'aide de ceux-ci, ils ont pu évaluer le degré de présence de codons spécifiques (biais de codon) pour des gènes individuels et pour des gènes hautement exprimés, la fréquence avec laquelle des codons particuliers étaient exprimés dans un gène, le biais de codon pour différentes espèces et le efficacité de l'interaction codon-anticodon. Cela a permis de déterminer le score de la paire de codons (CPS) dans les séquences de codage – «le logarithme naturel du rapport des observés sur le nombre attendu d'occurrences d'une paire de codons particulière dans toutes les séquences codant pour les protéines d'une espèce».
Pendant ce temps, le biais de la paire de codons a été utilisé pour trouver le CPS parmi les virus et les gènes hôtes. En d'autres termes, le nombre de fois où une paire de codons devrait se produire est une mesure du nombre de fois où il se produirait sans aucune association entre les codons de la paire. Une valeur CPS positive et négative montre qu'une paire de codons particulière est sur et sous-représentée dans la séquence d'intérêt.
Ainsi, le CPS a été calculé pour chacune des plus de 3 720 paires de codons possibles (61 x 61 codons). Les valeurs Enc ont été tracées par rapport aux valeurs GC3 pour montrer comment les mutations G + C ont affecté la relation entre elles, contrairement à l'effet de la pression de sélection. Des méthodes de regroupement ont été utilisées pour identifier les groupes de gènes à partir des similitudes dans l'utilisation des codons parmi les gènes humains et viraux.
L'analyse en composantes principales (ACP) a été effectuée pour trouver les facteurs les plus importants qui causent la variation entre les gènes. Enfin, ils ont réalisé une analyse phylogénétique sur les séquences du génome d'ADN de tous les virus pour dessiner un arbre phylogénétique.
En se basant sur le fait que les CoV humains ont un CUB qui correspond étroitement aux protéines hautement exprimées dans le tissu hôte infecté, les chercheurs ont examiné les molécules géniques de SARS-CoV-2 et MERS ainsi que les gènes humains. Ils ont constaté qu'au total, l'Enc moyenne était similaire parmi tous les gènes, viraux et humains, avec une seule unité de différence entre l'hôte non humain d'origine et le virus.
Correspondances du SARS-CoV-2 humain L'analyse a également montré que le SARS-CoV-2 humain était différent de celui des chauves-souris et des pangolins dans la distribution de certains gènes spécifiques, en fonction principalement de la teneur en A / T dans P3.
La distribution des gènes viraux importants pour la forme physique virale, comme la protéine M et la protéine E, partageait la tendance à un biais A / T et montrait une distribution différente de celle des virus non humains. Le CUB était plus élevé pour ces gènes par rapport au MERS humain et au SRAS, bien que cela contredit la théorie du compromis. Une autre explication serait l'effet de la pression de sélection qui favorise la réplication du virus dans un nouvel hôte, ou par le récent saut du virus à travers les frontières des espèces.
Implications de la correspondance des gènes humains et viraux
L'étude suggère donc que la réplication du virus chez l'homme est plus facile avec le regroupement de la protéine E avec des gènes humains qui présentent une correspondance moléculaire, ce qui facilite l'assemblage des virions et l'immunomodulation. Ceci est soutenu par le CPB positif et une corrélation CPS plus élevée pour le groupement de protéines E-gène humain.
Des schémas similaires sont observés avec d'autres gènes comme ORF6 et ORF8. Un CPB élevé est observé avec la protéine N, ORF1ab et la protéine S. Les changements dans la position GC3 conduisent à des substitutions synonymes et, à leur tour, à l'optimisation des codons dans les cellules humaines, en utilisant la machinerie des cellules hôtes pour traduire uniquement les gènes qui correspondent aux besoins viraux au niveau moléculaire.
De plus, cela pourrait conduire à une régulation négative des gènes humains dans le tissu pulmonaire, comme cela a été rapporté comme résultant de l'expression de l'ARNt déséquilibrée ou modifiée à tort. Ceci, à son tour, provoque une synthèse protéique incontrôlée ou anormale, produisant une maladie. Cela pourrait également expliquer certains effets d'une infection virale qui ne sont pas dus à une lésion virale directe.
L'étude conclut: «Dans nos études, nous avons fourni une liste de gènes humains qui pourraient être particulièrement affectés en raison de leurs similitudes moléculaires avec les gènes viraux, appartenant non seulement au SARS-CoV-2 mais également au SRAS et au MERS. Le dysfonctionnement de ces gènes a été associé à différentes pathologies humaines et est en augmentation continue. »
Cela pourrait aider à développer de nouveaux préventifs ainsi qu'à comprendre comment les gènes humains affectent la probabilité et les effets de COVID-19.
*Avis important
bioRxiv publie des rapports scientifiques préliminaires qui ne sont pas évalués par des pairs et, par conséquent, ne doivent pas être considérés comme concluants, guider la pratique clinique / les comportements liés à la santé, ou traités comme des informations établies.
















