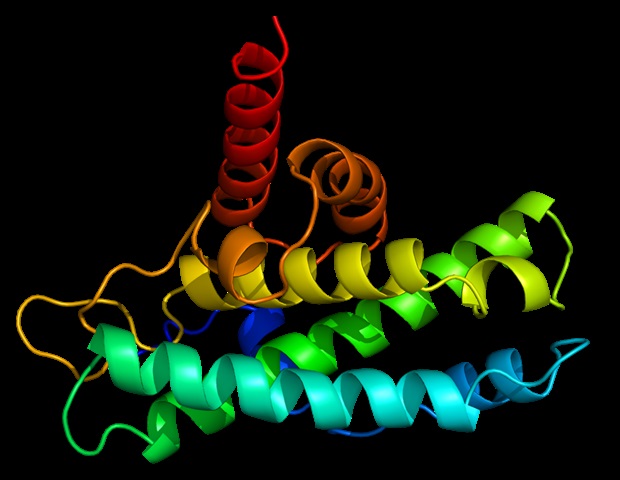
Lorsque vous pensez aux protéines – les enzymes, les molécules de signalisation et les composants structurels de tout être vivant – vous pensez peut-être à des brins simples d’acides aminés, organisés comme des perles sur une ficelle. Mais presque toutes les protéines sont constituées de plusieurs brins repliés et liés les uns aux autres, formant des superstructures 3D complexes appelées assemblages moléculaires. L’une des étapes clés pour comprendre la biologie consiste à découvrir comment une protéine fait son travail, ce qui nécessite une connaissance de ses structures jusqu’au niveau atomique.
Au cours du siècle dernier, les scientifiques ont développé et déployé des technologies étonnantes telles que la cristallographie aux rayons X et la microscopie cryoélectronique pour déterminer la structure des protéines, et ont ainsi répondu à d’innombrables questions importantes. Mais de nouveaux travaux montrent que comprendre la structure des protéines peut parfois être plus compliqué qu’on ne le pense.
Un groupe de chercheurs du Lawrence Berkeley National Laboratory (Berkeley Lab) étudiant la protéine la plus abondante au monde, une enzyme impliquée dans la photosynthèse appelée rubisco, a montré comment l’évolution peut conduire à une surprenante diversité d’assemblages moléculaires qui accomplissent tous la même tâche. Les conclusions, publiées aujourd’hui dans Avancées scientifiquesrévèlent la possibilité que de nombreuses protéines que nous pensions connaître existent en fait sous d’autres formes inconnues.
Historiquement, si les scientifiques résolvaient une structure et déterminaient qu’une protéine était dimère (composée de deux unités), par exemple, ils pouvaient supposer que des protéines similaires existaient également sous une forme dimère. Mais la petite taille de l’échantillon et le biais d’échantillonnage – des facteurs inévitables étant donné qu’il est très difficile de convertir des protéines naturellement liquides en formes solides et cristallisées qui peuvent être examinées par cristallographie aux rayons X – obscurcissaient la réalité.
« C’est comme si vous marchiez dehors et que vous voyiez quelqu’un promener son chien, si vous n’aviez jamais vu de chien auparavant, puis que vous voyiez un chien de saucisse, vous penseriez: » OK, c’est à quoi ressemblent tous les chiens. Mais ce que vous devez faire, c’est aller au parc canin et voir toute la diversité canine qui s’y trouve », a déclaré l’auteur principal Patrick Shih, chercheur dans le domaine des biosciences et directeur de la conception des biosystèmes végétaux au Joint BioEnergy Institute (JBEI). « Un point à retenir de cet article qui va au-delà du rubisco, à toutes les protéines, est la question de savoir si nous voyons ou non la véritable gamme de structures dans la nature, ou si ces biais donnent l’impression que tout ressemble à un chien de saucisse. »
Dans l’espoir d’explorer tous les différents arrangements de rubisco dans le parc à chiens métaphorique et d’apprendre d’où ils venaient, le laboratoire de Shih a collaboré avec des experts en biologie structurale de Bioscience Area en utilisant la source de lumière avancée de Berkeley Lab. Ensemble, l’équipe a étudié un type de rubisco (forme II) trouvé dans les bactéries et un sous-ensemble de microbes photosynthétiques en utilisant la cristallographie traditionnelle – une technique capable de résolution au niveau atomique – combinée à une autre technique de résolution de structure, la diffusion des rayons X aux petits angles. (SAXS), qui a une résolution inférieure mais peut prendre des instantanés de protéines dans leur forme native lorsqu’elles sont dans des mélanges liquides. SAXS a l’avantage supplémentaire d’une capacité à haut débit, ce qui signifie qu’il peut traiter des dizaines d’assemblages de protéines individuels en succession rapide.
Des travaux antérieurs avaient montré que le type de rubisco le mieux étudié trouvé dans les plantes (forme I) prend toujours un assemblage de «noyau octamérique» de huit grandes unités protéiques disposées avec huit petites unités, alors que la forme II était censée exister principalement sous forme de dimère avec un quelques rares exemples d’hexamères à six unités. Après avoir utilisé ces techniques complémentaires pour examiner des échantillons de rubisco provenant d’une gamme variée d’espèces de microbes, les auteurs ont observé que la plupart des protéines de rubisco de forme II sont en fait des hexamères, avec parfois des dimères, et ils ont découvert un tétramère (quatre unités) jamais vu auparavant. Assemblée.
La combinaison de ces données structurelles avec les séquences de gènes codant pour les protéines respectives a permis à l’équipe d’effectuer une reconstruction de séquence ancestrale – une méthode d’évolution moléculaire informatisée qui peut estimer à quoi ressemblaient les protéines ancestrales en fonction de la séquence et de l’apparence des protéines modernes qui en ont évolué.
La reconstruction suggère que le gène de la forme II rubisco a changé au cours de son histoire évolutive pour produire des protéines avec une gamme de structures qui se transforment en de nouvelles formes ou reviennent assez facilement à des structures plus anciennes. En revanche, au cours de l’évolution, des pressions sélectives ont conduit à une série de changements qui ont verrouillé la forme I rubisco en place – un processus appelé retranchement structurel – c’est pourquoi l’assemblage octamérique est le seul arrangement que nous voyons maintenant. Selon les auteurs, on a supposé que la plupart des assemblages de protéines étaient ancrés au fil du temps par une pression sélective pour affiner leur fonction, comme on le voit avec la forme I rubisco. Mais cette recherche suggère que l’évolution peut aussi favoriser les protéines flexibles.
« La grande découverte de cet article est qu’il y a beaucoup de plasticité structurelle », a déclaré Shih, qui est également professeur adjoint à l’UC Berkeley. « Les protéines peuvent être beaucoup plus flexibles, à travers le champ, que nous ne le pensions. »
Après avoir terminé la reconstruction de la séquence ancestrale, l’équipe a mené des expériences mutationnelles pour voir comment la modification de l’assemblage rubisco, dans ce cas la rupture d’un hexamère en un dimère, affectait l’activité de l’enzyme. De manière inattendue, cette mutation induite a produit une forme de rubisco qui utilise mieux sa molécule cible, le CO2. Tous les rubisco naturels se lient fréquemment à la molécule d’O2 de taille similaire par accident, ce qui réduit la productivité de l’enzyme. Il existe un grand intérêt pour la modification génétique du rubisco dans les espèces de plantes agricoles afin d’augmenter l’affinité de la protéine pour le CO2, afin de produire des cultures plus productives et économes en ressources. Cependant, on s’est beaucoup concentré sur le site actif de la protéine – la région de la protéine où le CO2 ou l’O2 se lient.
« C’est un aperçu intéressant pour nous car cela suggère que pour avoir des résultats d’ingénierie plus fructueux, nous ne pouvons pas simplement regarder la réponse la plus simple, la région de l’enzyme qui interagit réellement avec le CO2 », a déclaré le premier auteur Albert Liu. , un étudiant diplômé du laboratoire de Shih. « Peut-être qu’il y a des mutations en dehors de ce site actif qui participent réellement à cette activité et peuvent potentiellement modifier la fonction des protéines d’une manière que nous voulons. C’est donc quelque chose qui ouvre vraiment les portes à de futures voies de recherche. »
Le co-auteur Paul Adams, directeur associé du laboratoire pour les biosciences et vice-président pour la technologie chez JBEI, a ajouté : « Le mélange de techniques employées et la nature interdisciplinaire de l’équipe ont été une véritable clé du succès. Les travaux mettent en évidence la puissance de la combinaison des données génomiques et méthodes de biologie structurale pour étudier l’un des problèmes les plus importants de la biologie et parvenir à des conclusions inattendues. »